Super enhancer analysis
This post explores the most widely used super-enhancer identification tool ROSE.
And some errors I ran into in practice using ROSE.
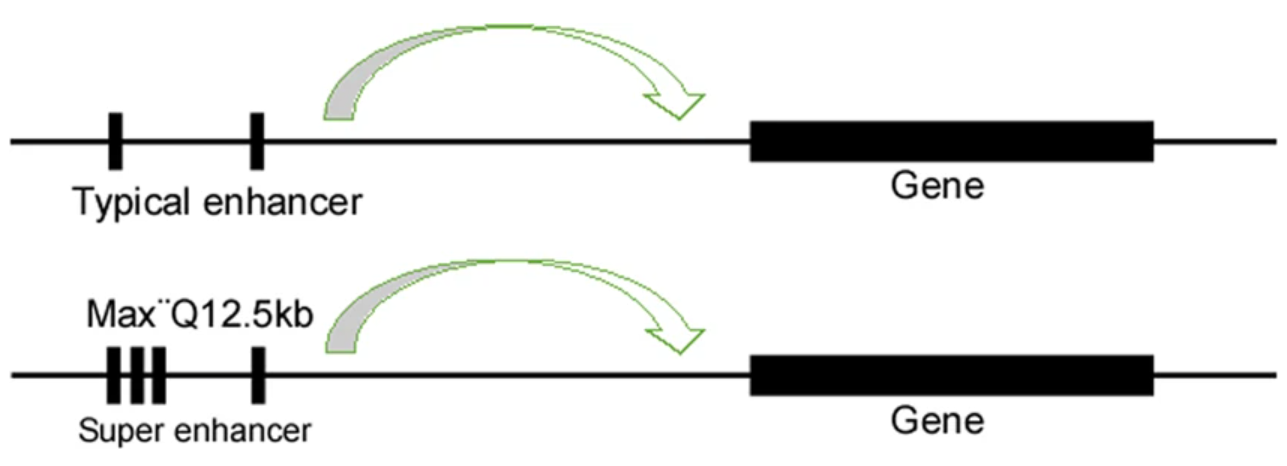
How does ROSE define super-enhancer?
- Combine the enhancers together that occur within 12.5kb of each other into a single larger enhancer region.
- Rank all enhancers by increasing total background-subtracted occupancy of enhancer markers (eg. H3K27ac). Meanwhile, plot the occupancy of in units of total rmp/bp (reads per million per base pair) in each ranking order, where you can see a clear break point where the occupancy signal starts rising rapidly.
- To find the break point, x and y axis are scaled to 0-1, and the point in the curve with a slope of 1 is regarded as the break point. The enhancers above it are super-enhancers, below it are typical enhancers.
How to use ROSE?
Simply download the software package from its official website and unzip it, all the scripts needed are included in the folder. A proper software environment with the right version of several tools is also essential running it: python (2.7.x), R (>2.15.3), samtools (>0.1.18).
Preparing input files:
- .gff file of enhancer regions previously identified, .gff files must have following columns:
1: chromosome (chr#)
2: unique ID for each constituent enhancer region
4: start of constituent
5: end of constituent
7: strand (+,-,.)
9: unique ID for each constituent enhancer region - .bam files of sequencing reads for factor of interest and control (WCE/IgG recommended)
Running:
1 | python ROSE_main.py -g hg19 -i enhancer.gff -r H3K27ac.bam -o SE_results |
Errors and solutions
- Indentation in raw script
TabError: inconsistent use of tabs and spaces in indentation
This error arises from the inconsistent indentation usage in some scripts you downloaded due to different editors. Solved by simply change the minor inconsistency.
- Python version
File “ROSE_main.py”, line 20, in <module>
from string import upper,join
ImportError: cannot import name ‘upper’ from ‘string’
This error showed up when I ignored the version of python I used which was 3.7.12. For ROSE, python of version 2.7.3 is a prerequisite, or functions in the same module might be different in other versions.
- Error in R graphic settings
Error in png(filename = plotFileName, height = 600, width = 600) :
X11 is not available
Execution halted
This error rose from the script ROSE_callSuper.R .
It was solved by reinstall the R in the conda environment.
Official tool website:
http://younglab.wi.mit.edu/super_enhancer_code.html
References:
Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes
Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers